

제품 전체 보호 아닌 승인 조건 단위 승인 제한 구조
특허 중심 경쟁에서 임상 설계 중심 전략 전환
 기자가 쓴 기사 더보기
기자가 쓴 기사 더보기





미국 식품의약국(FDA)이 3년 독점권 적용 기준을 구체화하면서, 제약바이오 업계의 전략 프레임이 특허 중심에서 임상 설계 중심으로 이동하고 있다. 기존에는 불명확했던 적용 범위와 해석 기준이 정리되면서, 동일 성분 의약품 간 경쟁 구도에도 변화가 불가피할 전망이다.
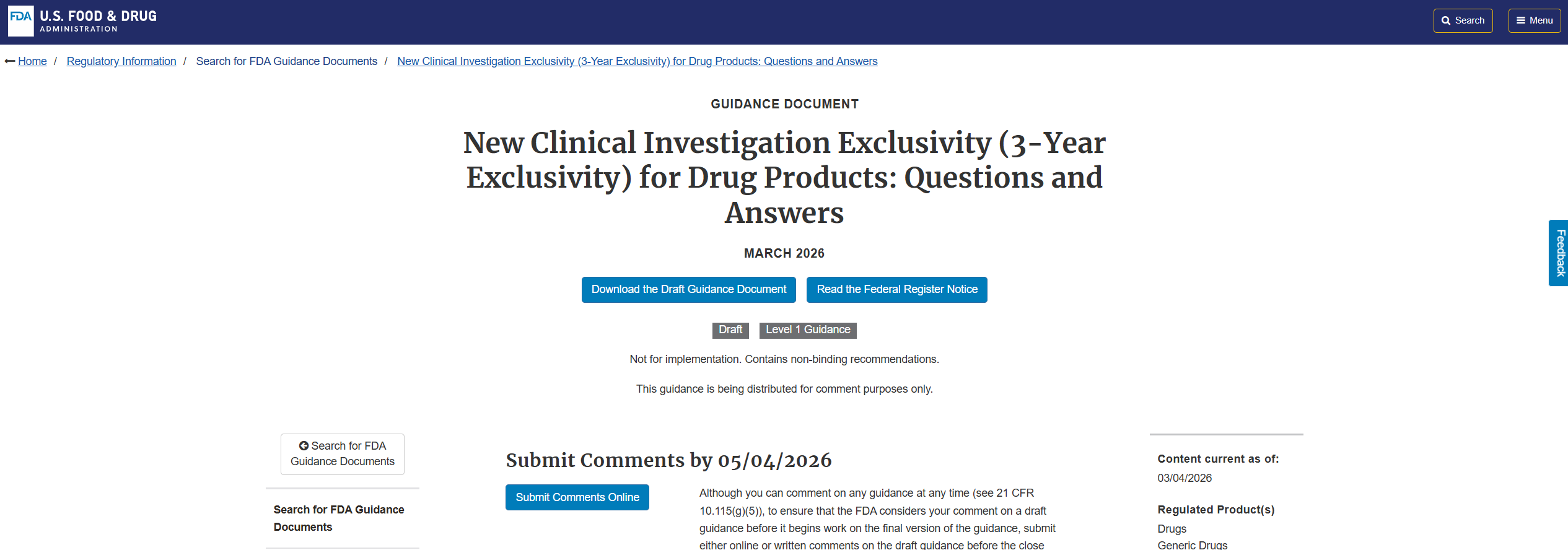
FDA는 최근 ‘신약 3년 독점권(New Clinical Investigation Exclusivity)’ 적용 기준을 Q&A 형식의 드래프트 가이던스로 공개하고, 지난 4일부터 Federal Register를 통해 공식 의견수렴 절차에 들어갔다.
해당 문서는 505(b)(2) 및 ANDA 승인 제한 범위, 임상시험 인정 요건(essential to approval), NDA 보충 신청 적용(NDA supplement) 가능성 등을 구체적으로 제시하며, 그동안 조문과 판례에 의존해 해석되던 기준을 처음으로 체계화했다.
법률사무소 WL LAW 이우진 변호사는 16일 ‘FDA 3년 독점권 초안 가이던스: 신규 임상시험 독점권(New Clinical Investigation Exclusivity)’ 보고서를 공식 블로그에 게재하고, 이번 가이던스의 핵심을 분석했다.
이 변호사는 “3년 독점권은 특허와 달리 제품 자체를 막는 제도가 아니라, 새 임상시험으로 확보한 승인 조건을 중심으로 경쟁사의 승인 시점을 조정하는 구조”라며 “어떤 임상 데이터를 확보하느냐에 따라 동일 성분이라도 시장 진입 구도가 달라질 수 있다”고 설명했다.
이어 그는 “이는 단순한 규제 해석 문제가 아니라 개발 전략의 문제”라며 “적응증 설정, 임상 설계, 승인 조건 구성까지 초기 단계부터 함께 설계해야 의미 있는 독점 효과를 만들 수 있다”고 강조했다.
바이오의약품은 대상 아니다…케미컬 중심 제도
3년 독점권은 모든 의약품에 적용되는 제도가 아니다. 연방식품의약품화장품법(FD&C Act) 505 조항에 근거한 제도로, NDA(신약승인신청) 또는 NDA 보충 신청 체계에 속한 의약품에 적용된다. 화학합성의약품, 즉 저분자(케미컬) 의약품이 주요 대상이다.
이 변호사는 “이번 가이던스는 FD&C Act 505 체계의 NDA 및 NDA 보충 신청을 전제로 한 설명으로, BLA 경로 생물의약품에 적용되는 별도 독점권 체계와는 구분해 해석할 필요가 있다”고 설명했다.
3년 독점권은 FD&C Act 505 체계의 NDA 및 NDA 보충 신청에 적용되는 제도로, BLA 경로 생물의약품에는 적용되지 않는다. 단일클론항체나 세포·유전자치료제 등 BLA 대상 생물의약품은 별도 12년 reference product exclusivity 체계를 적용받기 때문에 규제 구조 자체가 다르다.
경쟁사 판매 막는 제도 아닌, FDA 승인 제한
또 하나의 핵심은 제한 대상이다. 3년 독점권은 경쟁사 제품 판매를 직접 금지하는 제도가 아니다. 기업의 상업 행위를 제한하는 것이 아닌, FDA 승인 행위를 제한하는 구조로 봐야한다.
법적으로는 FDA가 경쟁사의 후속 신청을 승인하는 것이 제한된다. 보다 정확히는, 경쟁사 505(b)(2) 신청 또는 ANDA에 대해 ‘독점권으로 보호되는 승인 조건’에 관한 승인을 FDA가 내릴 수 없도록 하는 제도다. 동일한 제품이라도 보호 대상이 아닌 조건에 대해서는 승인 자체가 가능하다.
이 변호사에 따르면, 3년 독점권은 경쟁사의 시장 진입 자체를 금지하는 것이 아니라 FDA의 승인 권한을 특정 승인 조건에 대해 제한하는 제도로 작동한다. 특허처럼 직접적인 배타권이 아니라, 경쟁사 승인 시점을 지연시키는 규제 장치라는 설명이다.
이 구조 때문에 경쟁사는 개발·생산·허가 신청까지는 모두 진행할 수 있지만, 특정 조건이 포함된 형태로는 FDA 승인을 받을 수 없다. 다시 말해, 시장 진입 자체가 막히는 것이 아니라, 어떤 형태로 언제 진입할 수 있는지가 제한된다.
예를 들어 특정 적응증이나 용법·용량을 새로운 임상시험으로 승인받아 3년 독점권을 확보한 경우, 경쟁사는 동일 조건을 포함한 신청을 제출할 수 있다. 그러나 FDA는 해당 조건에 대해 승인을 내릴 수 없다. 결과적으로 경쟁사는 해당 조건을 제외한 형태로 신청을 구성하거나, 독점 기간 종료 이후를 기다려야 한다.
제품 전체 아닌 ‘승인 조건’ 단위 보호
3년 독점권은 제품 전체를 보호하지 않는다. 보호 범위는 exclusivity-protected conditions of approval, 즉 새 임상시험으로 입증된 승인 조건에 한정된다.
이 변호사는 “3년 독점권은 제품 전체가 아니라, 새 임상시험으로 입증된 승인 조건에 대해서만 보호 효과가 발생한다는 점이 핵심”이라며 “적응증, 용법·용량, 투여 경로, 환자군 등 승인 요소별로 보호 범위가 나뉜다”고 설명했다.
기존 약물에 소아 적응증을 추가 승인받았다면, FDA는 경쟁사의 신청 중 소아 적응증 부분에 대해서만 승인을 제한한다. 반면, 기존에 승인돼 있던 성인 적응증에 대해서는 경쟁사의 승인을 제한하지 않는다.
PK 연구만으로 부족…‘승인 기여도’가 판단 기준
임상시험 요건도 명확하다. FDA는 PK(약동학) 또는 생체이용률 확인만을 목적으로 하는 연구는 원칙적으로 3년 독점권을 위한 임상시험으로 보기 어렵다고 설명했다.
핵심은 해당 연구가 ‘승인에 필수적(essential to approval)’이었는지 여부다. 해당 데이터가 없으면 승인 자체가 어려웠다고 판단될 수준이어야 한다는 것.
이 변호사는 “단순히 임상시험을 수행했다는 사실만으로는 독점권이 인정되지 않고, 해당 데이터가 승인 판단에 필수적이었는지가 기준이 된다”며 “형식이 아니라 승인 기여도가 판단의 핵심”이라고 설명했다.
다만 안전성·내약성 평가가 1차 평가변수로 설정되고, 그 결과가 승인 근거로 활용된 경우라면 PK 요소가 포함돼 있어도 임상시험으로 인정될 수 있다. 결국 판단 기준은 연구 형식이 아니라 승인 기여도다.
보충 신청도 가능…그러나 변경 범위만 적용
NDA 보충 신청을 통해서도 3년 독점권을 확보할 수 있다. 적용 범위는 제한적이다. 기존 승인 조건 전체는 받을 수 없고, 새 임상시험으로 입증된 변경된 승인 조건에 대해서만 독점권이 발생한다.
적용 대상은 적응증 추가, 용량·용법 변경, 투여 경로 변경, 특정 환자군 확대 등 명확히 구분되는 승인 변경 사항이다. 이때 해당 임상시험도 ‘승인에 필수적’으로 인정돼야 한다.
경쟁사는 기존 승인 조건을 기반으로 한 신청은 가능하다. 그러나 보충 신청으로 추가된 적응증이나 변경된 조건을 포함한 신청에 대해서는 해당 기간 동안 FDA 승인을 받을 수 없다. 대신 해당 조건을 제외하는 ‘carve-out’ 방식은 허용될 수 있어, 실제 시장 차단 범위는 승인 조건 구성에 따라 달라진다.
결국 보충 신청 기반 3년 독점권은 제품 전체를 재보호하는 제도가 아니다. 특정 승인 조건 단위로 경쟁사 진입 시점을 지연시키는 제한적 보호 장치에 가깝다.
이 변호사는 “3년 독점권은 특허와 별도로 작동하는 규제 수단인 만큼, 임상 개발 초기 단계부터 적용 가능성을 전략적으로 검토해야 한다”면서 “특히 신규 임상시험 해당 여부와 기존 데이터와의 구분, 유효성 엔드포인트의 승인 기여도, 다중 코호트 설계 시 사전 명시 요건 등을 충족하도록 프로토콜을 설계하는 것이 중요하다”고 설명했다.
이어 그는 “독점권 확보 여부는 NDA 제출 단계에서 결정되는 것이 아니라 개발 초기부터 준비된 문서 체계와 임상 설계에 의해 좌우된다”라며 “21 CFR 314.50(j)에 따른 독점권 신청 전략까지 포함해 사전에 통합적으로 설계해야 실질적인 시장 보호 효과를 확보할 수 있다”고 강조했다.
한편 이번 가이던스 ‘New Clinical Investigation Exclusivity (3-Year Exclusivity) for Drug Products: Questions and Answers Guidance for Industry’는 FDA 공식 홈페이지에서 확인할 수 있다.






| 인기기사 | 더보기 + |
-
1 미국, 중국 바이오 투자·라이선싱까지 조인다…K바이오 CDMO 반사이익 생기나 -
2 [제약 AI 대전환①] "사용량은 급증, 업무 성과는 제로?"… 헛도는 제약사 생성형 AI -
3 [스페셜리포트]인제니아테라퓨틱스, MSD 후기 임상 오른 'IGT-427'…TIE2 기술 가치 확장 -
4 mRNA 독감백신 허가…‘엠플루시바’ 80만명 검증대 오른다 -
5 에이비온, iRAC™ 플랫폼 원천기술 미국 특허 등록 -
6 제품 개발 문법 된 K-뷰티…글로벌 기업 '리버스 듀프' 뚜렷 -
7 아리바이오, 아리바이오랩 누적 50억원 투자..지분·R&D 협력 강화 -
8 리브스메드, K-수술로봇 '스타크' 식약처 품목허가 신청 -
9 후천성 시상하부 비만 신약 임상 2상 긍정 시그널 -
10 올릭스,RNAi 개발 전문가 민지영 박사 수석부사장 겸 CDO 영입
| 인터뷰 | 더보기 + |
| PEOPLE | 더보기 + |
| 컬쳐/클래시그널 | 더보기 + |
|
|
서울시 서초구 서초대로 115 정다운빌딩 4층 TEL : 02-3270-0114 FAX : 02-3270-0189 E-mail : webmaster@yakup.com |
발행일 : 1954. 03. 29 등록일 : 2016. 05. 02 대표이사 : 함태원 발행 · 편집인 : 함태원 등록번호 : 서울,아04071 |
사업자등록번호 : 106-81-10940 통신사업자신고번호 : 제2022-서울서초-2586호 청소년보호책임자 : 이종운
Copyright © Yakup.com All rights reserved.
|

- 권혁진 기자 hjkwon@yakup.com
- 입력 2026.03.18 06:00 수정 2026.03.18 06:06
미국 식품의약국(FDA)이 3년 독점권 적용 기준을 구체화하면서, 제약바이오 업계의 전략 프레임이 특허 중심에서 임상 설계 중심으로 이동하고 있다. 기존에는 불명확했던 적용 범위와 해석 기준이 정리되면서, 동일 성분 의약품 간 경쟁 구도에도 변화가 불가피할 전망이다.
FDA는 최근 ‘신약 3년 독점권(New Clinical Investigation Exclusivity)’ 적용 기준을 Q&A 형식의 드래프트 가이던스로 공개하고, 지난 4일부터 Federal Register를 통해 공식 의견수렴 절차에 들어갔다.
해당 문서는 505(b)(2) 및 ANDA 승인 제한 범위, 임상시험 인정 요건(essential to approval), NDA 보충 신청 적용(NDA supplement) 가능성 등을 구체적으로 제시하며, 그동안 조문과 판례에 의존해 해석되던 기준을 처음으로 체계화했다.
법률사무소 WL LAW 이우진 변호사는 16일 ‘FDA 3년 독점권 초안 가이던스: 신규 임상시험 독점권(New Clinical Investigation Exclusivity)’ 보고서를 공식 블로그에 게재하고, 이번 가이던스의 핵심을 분석했다.
이 변호사는 “3년 독점권은 특허와 달리 제품 자체를 막는 제도가 아니라, 새 임상시험으로 확보한 승인 조건을 중심으로 경쟁사의 승인 시점을 조정하는 구조”라며 “어떤 임상 데이터를 확보하느냐에 따라 동일 성분이라도 시장 진입 구도가 달라질 수 있다”고 설명했다.
이어 그는 “이는 단순한 규제 해석 문제가 아니라 개발 전략의 문제”라며 “적응증 설정, 임상 설계, 승인 조건 구성까지 초기 단계부터 함께 설계해야 의미 있는 독점 효과를 만들 수 있다”고 강조했다.
바이오의약품은 대상 아니다…케미컬 중심 제도
3년 독점권은 모든 의약품에 적용되는 제도가 아니다. 연방식품의약품화장품법(FD&C Act) 505 조항에 근거한 제도로, NDA(신약승인신청) 또는 NDA 보충 신청 체계에 속한 의약품에 적용된다. 화학합성의약품, 즉 저분자(케미컬) 의약품이 주요 대상이다.
이 변호사는 “이번 가이던스는 FD&C Act 505 체계의 NDA 및 NDA 보충 신청을 전제로 한 설명으로, BLA 경로 생물의약품에 적용되는 별도 독점권 체계와는 구분해 해석할 필요가 있다”고 설명했다.
3년 독점권은 FD&C Act 505 체계의 NDA 및 NDA 보충 신청에 적용되는 제도로, BLA 경로 생물의약품에는 적용되지 않는다. 단일클론항체나 세포·유전자치료제 등 BLA 대상 생물의약품은 별도 12년 reference product exclusivity 체계를 적용받기 때문에 규제 구조 자체가 다르다.
경쟁사 판매 막는 제도 아닌, FDA 승인 제한
또 하나의 핵심은 제한 대상이다. 3년 독점권은 경쟁사 제품 판매를 직접 금지하는 제도가 아니다. 기업의 상업 행위를 제한하는 것이 아닌, FDA 승인 행위를 제한하는 구조로 봐야한다.
법적으로는 FDA가 경쟁사의 후속 신청을 승인하는 것이 제한된다. 보다 정확히는, 경쟁사 505(b)(2) 신청 또는 ANDA에 대해 ‘독점권으로 보호되는 승인 조건’에 관한 승인을 FDA가 내릴 수 없도록 하는 제도다. 동일한 제품이라도 보호 대상이 아닌 조건에 대해서는 승인 자체가 가능하다.
이 변호사에 따르면, 3년 독점권은 경쟁사의 시장 진입 자체를 금지하는 것이 아니라 FDA의 승인 권한을 특정 승인 조건에 대해 제한하는 제도로 작동한다. 특허처럼 직접적인 배타권이 아니라, 경쟁사 승인 시점을 지연시키는 규제 장치라는 설명이다.
이 구조 때문에 경쟁사는 개발·생산·허가 신청까지는 모두 진행할 수 있지만, 특정 조건이 포함된 형태로는 FDA 승인을 받을 수 없다. 다시 말해, 시장 진입 자체가 막히는 것이 아니라, 어떤 형태로 언제 진입할 수 있는지가 제한된다.
예를 들어 특정 적응증이나 용법·용량을 새로운 임상시험으로 승인받아 3년 독점권을 확보한 경우, 경쟁사는 동일 조건을 포함한 신청을 제출할 수 있다. 그러나 FDA는 해당 조건에 대해 승인을 내릴 수 없다. 결과적으로 경쟁사는 해당 조건을 제외한 형태로 신청을 구성하거나, 독점 기간 종료 이후를 기다려야 한다.
제품 전체 아닌 ‘승인 조건’ 단위 보호
3년 독점권은 제품 전체를 보호하지 않는다. 보호 범위는 exclusivity-protected conditions of approval, 즉 새 임상시험으로 입증된 승인 조건에 한정된다.
이 변호사는 “3년 독점권은 제품 전체가 아니라, 새 임상시험으로 입증된 승인 조건에 대해서만 보호 효과가 발생한다는 점이 핵심”이라며 “적응증, 용법·용량, 투여 경로, 환자군 등 승인 요소별로 보호 범위가 나뉜다”고 설명했다.
기존 약물에 소아 적응증을 추가 승인받았다면, FDA는 경쟁사의 신청 중 소아 적응증 부분에 대해서만 승인을 제한한다. 반면, 기존에 승인돼 있던 성인 적응증에 대해서는 경쟁사의 승인을 제한하지 않는다.
PK 연구만으로 부족…‘승인 기여도’가 판단 기준
임상시험 요건도 명확하다. FDA는 PK(약동학) 또는 생체이용률 확인만을 목적으로 하는 연구는 원칙적으로 3년 독점권을 위한 임상시험으로 보기 어렵다고 설명했다.
핵심은 해당 연구가 ‘승인에 필수적(essential to approval)’이었는지 여부다. 해당 데이터가 없으면 승인 자체가 어려웠다고 판단될 수준이어야 한다는 것.
이 변호사는 “단순히 임상시험을 수행했다는 사실만으로는 독점권이 인정되지 않고, 해당 데이터가 승인 판단에 필수적이었는지가 기준이 된다”며 “형식이 아니라 승인 기여도가 판단의 핵심”이라고 설명했다.
다만 안전성·내약성 평가가 1차 평가변수로 설정되고, 그 결과가 승인 근거로 활용된 경우라면 PK 요소가 포함돼 있어도 임상시험으로 인정될 수 있다. 결국 판단 기준은 연구 형식이 아니라 승인 기여도다.
보충 신청도 가능…그러나 변경 범위만 적용
NDA 보충 신청을 통해서도 3년 독점권을 확보할 수 있다. 적용 범위는 제한적이다. 기존 승인 조건 전체는 받을 수 없고, 새 임상시험으로 입증된 변경된 승인 조건에 대해서만 독점권이 발생한다.
적용 대상은 적응증 추가, 용량·용법 변경, 투여 경로 변경, 특정 환자군 확대 등 명확히 구분되는 승인 변경 사항이다. 이때 해당 임상시험도 ‘승인에 필수적’으로 인정돼야 한다.
경쟁사는 기존 승인 조건을 기반으로 한 신청은 가능하다. 그러나 보충 신청으로 추가된 적응증이나 변경된 조건을 포함한 신청에 대해서는 해당 기간 동안 FDA 승인을 받을 수 없다. 대신 해당 조건을 제외하는 ‘carve-out’ 방식은 허용될 수 있어, 실제 시장 차단 범위는 승인 조건 구성에 따라 달라진다.
결국 보충 신청 기반 3년 독점권은 제품 전체를 재보호하는 제도가 아니다. 특정 승인 조건 단위로 경쟁사 진입 시점을 지연시키는 제한적 보호 장치에 가깝다.
이 변호사는 “3년 독점권은 특허와 별도로 작동하는 규제 수단인 만큼, 임상 개발 초기 단계부터 적용 가능성을 전략적으로 검토해야 한다”면서 “특히 신규 임상시험 해당 여부와 기존 데이터와의 구분, 유효성 엔드포인트의 승인 기여도, 다중 코호트 설계 시 사전 명시 요건 등을 충족하도록 프로토콜을 설계하는 것이 중요하다”고 설명했다.
이어 그는 “독점권 확보 여부는 NDA 제출 단계에서 결정되는 것이 아니라 개발 초기부터 준비된 문서 체계와 임상 설계에 의해 좌우된다”라며 “21 CFR 314.50(j)에 따른 독점권 신청 전략까지 포함해 사전에 통합적으로 설계해야 실질적인 시장 보호 효과를 확보할 수 있다”고 강조했다.
한편 이번 가이던스 ‘New Clinical Investigation Exclusivity (3-Year Exclusivity) for Drug Products: Questions and Answers Guidance for Industry’는 FDA 공식 홈페이지에서 확인할 수 있다.
무단 전재·복사·배포 등을 금지합니다.