

FDA, 위험 기반·생애주기 중심 심사 원칙 강조
일본 조건·기한부 승인제도 운영 현황 공개
중국 가속 승인·조건부 허가 제도 확대
식약처, 비교동등성 평가 가이드라인 핵심 제시
 기자가 쓴 기사 더보기
기자가 쓴 기사 더보기





세포·유전자치료제(Cell & Gene Therapy, CGT)가 글로벌 의약품 개발의 핵심 축으로 부상하면서, 각국 규제기관은 기존 의약품 규제 틀을 유지하면서도 첨단 치료제의 특성을 반영한 제도적 진화를 동시에 모색하고 있다. 생물학적 복잡성, 맞춤형 제조공정, 소규모·희귀질환 환자군 대상 임상 설계, 장기추적 안전성 관리 등 CGT 고유의 특성은 허가 심사, 사후 관리, 국제 조화 전략 전반에 걸쳐 새로운 기준 정립을 요구하고 있다. 이에 따라 주요 규제기관들은 심사체계 단순화, 가속 승인 프로그램 확대, 위험 기반 접근, 비교성·비교동등성 평가 고도화, 실제임상자료 활용 등 다양한 정책 수단을 통해 개발 촉진과 환자 보호 간 균형을 재정립하는 방향으로 제도를 발전시키고 있다.
이 같은 흐름을 한 자리에서 조망하는 자리가 마련됐다. Drug Information Association(DIA)는 지난 27일 서울 세브란스병원 유일한 기념홀에서 ‘Pioneering Precision: Cell and Gene Therapy Development and Innovation’을 주제로 ‘DIA Cell and Gene Therapy Summit’을 개최했다.
이날 세션1 ‘Regulatory Overview of Cell and Gene Therapy Field’에서는 ▲Caroline Pothet, Advanced Therapies and Haemato-Oncology Head(European Medicines Agency(EMA)), ▲Tamei Elliott, DIA US Director of Global Scientific Content(U.S. Food and Drug Administration(FDA)), ▲Yasuhiro Kishioka(Review Director, Office of Cellular and Tissue-based Products(Pharmaceuticals and Medical Devices Agency(PMDA)), ▲Jinanchao Gao, Associate Researcher Changping Laboratory(National Medical Products Administration(NMPA)), ▲왕소영 식품의약품안전처 식품의약품안전평가원 세포유전자치료제과장(식품의약품안전처) 등 주요 규제기관 관계자들이 참여해 자국의 CGT 규제체계 현황과 최근 제도 개편 동향을 발표했다.
발표는 유럽의 ATMP 법제 개편과 위원회 구조 단순화, 미국의 위험 기반·생애주기 중심 심사 접근, 일본의 조건·기한부 승인제도와 이원화된 법체계, 중국의 가속 승인 프로그램과 조건부 허가 운영, 한국의 첨단재생바이오법 기반 전주기 관리체계와 비교동등성 평가 가이드라인 등으로 이어졌다.
각 기관은 허가 심사 효율화와 조기 접근성 확보, 제조 변경에 따른 비교성 관리, 사후 임상근거 축적, 국제 조화 활동 등 공통 과제를 공유하면서도, 자국 제도에 기반한 차별화된 정책 도구를 제시했다. 이번 세션은 CGT 시대를 맞아 글로벌 규제 환경이 어떠한 방향으로 정비되고 있는지를 종합적으로 보여주는 자리로 마련됐다.

EMA
Caroline Pothet, 유럽의약품청(EMA) Advanced Therapies and Haemato-Oncology Head는 ‘Regulatory Update from the EMA’를 주제로 유럽 첨단치료제(ATMP) 규제 체계의 현황과 향후 개편 방향을 설명했다.
발표에 따르면, 유럽 ATMP 규제의 근간은 2007년 제정돼 2009년 발효된 ATMP Regulation이다. 이 법은 특정 제품군에 적용되는 특별법(Lex specialis)으로, ATMP의 정의와 범위를 명확히 하고 개발사가 자사 제품의 ATMP 해당 여부에 대해 사전 분류(classification)를 요청할 수 있는 제도를 도입했다.
또한 첨단치료제위원회(CAT)를 설치하고 ATMP에 대해 중앙집중 허가절차(centralised procedure)를 의무화했으며, 품질·유효성·안전성 및 긍정적 이익-위험 평가 입증을 허가 요건으로 규정했다. 허가 후 추가 유효성 및 안전성 자료 제출 요구 근거도 마련했으며, 중소기업을 위한 ATMP 인증(certification) 제도도 포함했다.
ATMP는 크게 ▲유전자치료제(Gene Therapy Medicinal Products) ▲체세포치료제(Somatic Cell Therapy Medicinal Products) ▲조직공학제제(Tissue Engineered Products) ▲의료기기와 결합된 복합 ATMP로 구분된다.
유전자치료제는 재조합 핵산을 포함해 유전적 변화를 유도하는 제품이며, 체세포치료제와 조직공학제제는 실질적 조작(substantial manipulation)을 거친 세포·조직을 활용해 질환 치료 또는 조직 재생·대체를 목표로 한다.
Caroline Pothet은 2023년 12월 유럽연합 집행위원회, 이사회, 유럽의회가 정치적 합의에 도달한 EU 신 의약품 법제(New Pharmaceutical Legislation, NPL)에 대해서도 설명했다. 해당 법제는 2028년부터 적용될 예정이며, 환자 접근성 강화, 경쟁력 및 혁신 지원, 미래 위기 대비, 의약품 공급 안정화 등 네 가지 축을 중심으로 구성된다.
ATMP와 관련해 가장 주목되는 변화는 유전자치료제 정의의 확대다. 기존 ‘recombinant nucleic acid’ 중심 정의에서 ‘synthetic nucleic acid’까지 범위를 넓히고, 유전자편집(gene editing) 기능을 명시적으로 포함할 예정이다.
허가 체계 측면에서는 위원회 구조가 단순화된다. 현재 CAT, CHMP, PRAC, PDCO, COMP 등 복수 위원회가 관여하는 구조에서 향후 CHMP와 PRAC 중심의 체계로 재편된다. 이에 따라 ATMP도 CHMP가 직접 평가를 담당하며, PRAC은 안전성 및 약물감시를 지속적으로 담당한다. 소아개발계획(PIP) 및 희귀의약품 관련 기능은 EMA와 CHMP가 분담하는 구조로 조정된다.
평가기간도 기존 210일에서 180일로 단축되며, 허가 갱신 절차 없이 무기한 허가가 부여된다. 또한 전자 제출 의무화, 임상시험 데이터의 전자적 제출, 전자 제품정보(electronic product information) 도입 등이 포함된다.
임상적 가치 평가와 관련해서는 2025년 1월부터 ATMP 및 항암제를 대상으로 EU 차원의 공동 임상평가(Joint Clinical Assessment, JCA)가 의무화됐다. 이는 허가 심사와 병행해 HTA 기관과의 조율을 진행하는 구조로, 이후 적용 범위가 점진적으로 확대될 예정이다.
ATMP Regulation의 특징 중 하나인 병원면제(Hospital Exemption) 제도에 대해서도 개편이 예고됐다. 국가별 관리 편차를 줄이기 위해 병원면제 제품의 공공 등록 의무화, 사용 의사의 유효성·안전성 데이터 수집 의무, 회원국 감독 강화 등이 도입될 예정이다.
혁신 지원 측면에서는 과학자문 체계 확대, HTA 기관과의 공동 절차 도입, PRIME 제도의 법제화 및 조기 지원 강화, 규제 샌드박스(Regulatory Sandbox) 도입, 소아개발계획의 연속적 업데이트 허용 등이 포함된다.
또한 ATMP 분류 제도는 유지되며, 향후에는 해당 제품이 의약품에 해당하는지 여부에 대해서도 EMA가 과학적 의견을 제시할 수 있게 된다. 의료기기·식품 등 타 규제 체계와의 협의도 가능하도록 구조가 조정된다.
Caroline Pothet은 이번 개편이 규제 간소화와 절차 효율화, 유전자치료 정의 확대, 병원면제 감독 강화, 디지털 전환 가속화 등을 포함하는 전면적 구조 개편이라고 설명하며 발표를 마무리했다.

FDA
Tamei Elliott(DIA US Director of Global Scientific Content)는 ‘FDA Update’를 주제로 미국 식품의약국(FDA)의 세포·유전자치료제 규제 체계와 최근 정책 방향을 설명했다.
발표에 따르면, 세포·유전자치료제의 기술적 특성과 개발 환경 변화는 규제 과학의 진화를 촉진하고 있다. 플랫폼 기술의 빠른 발전, 소규모·희귀질환 환자군을 대상으로 한 치료제 개발 확대, 복잡한 제조공정과 빈번한 생애주기 변경, 장기 안전성 모니터링 의무 등은 기존 규제 접근 방식에 보완을 요구하고 있다는 설명이다. 이에 따라 FDA는 혁신 촉진과 환자 보호 사이의 균형을 유지하는 방향으로 규제 적용 방식을 조정하고 있다고 밝혔다.
미국에서 대부분의 세포·유전자치료제는 공중보건서비스법(Public Health Service Act) 제351조에 따른 생물학적 제제로 규제된다. 이들 제품은 안전성, 순도, 효력에 대한 전면적 사전 심사를 거쳐야 하며, 개발은 임상시험계획(IND) 제출로 시작해 임상시험을 진행한 후 생물학적제제허가신청(BLA)으로 이어진다.
Tamei Elliott은 법적 기반과 핵심 요건 자체는 유지되고 있으며, 세포·유전자치료제라고 해서 품질·안전성·유효성에 대한 기본 의무가 완화되는 것은 아니라고 설명했다.
다만 FDA는 이러한 기존 규정을 세포·유전자치료제의 특성에 맞게 적용하는 방식에서 진화를 보이고 있다. 근거 창출 전략, 제조 및 품질관리(CMC), 비교성 관리, 효력 시험 전략 등에서 기대 수준이 구체화되고 있으며, 특히 생애주기 관점에서의 사전 계획을 강조하고 있다. 제조 변경이나 공정 개선을 개발 후반에 대응적으로 처리하기보다, 초기 단계부터 비교성 전략을 설계할 것을 요구하고 있다는 설명이다.
FDA는 세포·유전자치료제와 관련한 다양한 가이드라인을 제시하고 있으며, 이는 임상개발, 첨단 치료기술, CMC 및 품질, 프로그램 정책 등을 포괄한다. 가이드라인은 법적 구속력을 갖는 규정은 아니지만, 해당 분야에 대한 FDA의 현재 사고방식을 반영하는 문서로 활용된다. 소규모 환자군을 대상으로 한 임상시험 설계와 관련해서는 단일군 시험에서 피험자를 자기 대조군으로 활용하는 방식, 질병 진행 모델링, 외부 대조군 연구, 적응적 설계, 베이지안 접근, 마스터 프로토콜 등 다양한 혁신적 설계 방안이 논의되고 있다.
허가 후 안전성 및 유효성 자료 수집과 관련해서는 실제임상자료 및 실제임상근거 활용, 환자등록시스템 구축, 전자의무기록, 의료 청구자료, 생명통계자료 등 다양한 데이터원을 활용하는 방안이 제시됐다. 이는 장기 추적관찰이 필요한 세포·유전자치료제의 특성을 반영한 접근이다.
발표에서는 중증 질환을 대상으로 하는 치료제에 적용 가능한 신속 프로그램도 소개됐다. Fast Track, Breakthrough Therapy, Regenerative Medicine Advanced Therapy(RMAT), Priority Review, Accelerated Approval 등이 포함되며, 재생의학 치료는 세포치료제, 조직공학 제품, 인체세포·조직 제품, 관련 복합제품 등으로 정의된다.
최근 FDA는 세포·유전자치료제에 대해 위험 기반·과학 기반 유연성을 강조하고 있다. 다만 유연성은 규제 수준의 완화를 의미하지 않으며, 제품 위험도와 과학적 근거에 따라 개별적으로 적용된다. 예로는 임상 2·3상 이전 단계에서 상업용 완전 cGMP 요건을 일괄적으로 요구하지 않는 점, 공정 및 방법 검증에 대한 생애주기 접근, 공정적격성평가(PPQ) 배치 수의 유연한 적용, 소규모 환자군 대상 출하 기준의 탄력적 설정, 제조 경험에 따른 허가 후 기준 조정 등이 제시됐다.
FDA는 개발 초기 단계에서의 조기 소통을 지속적으로 권장하고 있으며, Pre-IND, End-of-Phase 2, Pre-Phase 3, BLA 단계 등에서 공식 회의(Type meeting)를 운영하고 있다. 서면 요청 시 60일 이내 회의를 개최해야 하는 절차가 마련돼 있다. 또한 라운드테이블과 워크숍을 통해 개발 과정의 도전과제를 파악하고 이를 향후 가이드라인과 심사 접근에 반영하고 있다고 설명했다.
최근 심사 환경과 관련해서는 문서화 수준 강화, 비교성 전략 및 평가변수의 타당성 확보, 비공식적 합의에 의존하지 않는 전략 수립의 필요성이 언급됐다.
Tamei Elliott은 세포·유전자치료제 개발 시 FDA의 생애주기 기대 수준을 반영해 초기 단계부터 규제 전략을 설계해야 하며, 가이드라인의 변화는 향후 심사 방향을 가늠할 수 있는 신호로 해석할 필요가 있다고 정리했다.

PMDA
Yasuhiro Kishioka(Review Director, Office of Cellular and Tissue-based Products, PMDA)는 일본의 재생의료제품 규제체계와 최근 승인 동향, 개발 촉진 제도 및 국제조화 활동을 중심으로 발표했다.
발표에 따르면, 일본은 2014년 재생의료에 대한 기대 증가를 배경으로 두 개의 법 체계를 정비했다. 하나는 ‘재생의료 등의 안전성 확보 등에 관한 법률(Safety Act)’이며, 다른 하나는 약사법을 개정한 ‘의약품의료기기등법(PMD Act)’이다.
Safety Act는 의료기관에서 시행되는 의료행위 및 비상업적 임상연구를 규율하며, 후생노동성이 관할한다. 반면 PMD Act는 재생의료제품의 임상시험계획 신고, 허가 심사 및 과학적 자문을 포함한 규제 절차를 규율하며, PMDA가 심사를 담당한다.
PMD Act상 재생의료제품은 세포기반 제품, 조직기반 제품, 유전자치료제를 포함하는 개념으로 정의된다.
Yasuhiro Kishioka는 ATMP와 유사한 범주로 이해할 수 있으나, 일본 법체계에 근거한 독자적 정의가 적용된다고 설명했다.
현재 일본에서 PMD Act에 따라 승인된 재생의료제품은 다수에 이르며, 적응증별로는 종양 분야 제품 비중이 높은 것으로 나타났다. 체세포치료제와 줄기세포 기반 제품도 다양한 적응증에서 허가를 받았다. 최근에는 iPS 세포 유래 제품 2건이 후생노동성 자문기구에서 조건부 승인을 권고받았다고 언급됐다.
일본은 재생의료제품 개발 촉진을 위해 오픈지정제도(Orphan designation), 사키가케(Sakigake) 지정, 조건·기한부 승인제도(Conditional and Time-limited Approval) 등 세 가지 제도를 운영하고 있다. 전체 승인 제품 중 80% 이상이 희귀의약품으로 지정됐으며, 지정 시 우선 과학적 자문, 우선 심사 등의 인센티브가 제공된다. 일반적인 허가 심사기간은 12개월이지만, 희귀 지정 제품의 경우 9개월로 단축된다.
사키가케 지정은 혁신 제품의 조기 도입을 목적으로 하며, 일본에서 최초 또는 글로벌과 동시 개발을 추진하는 경우를 요건으로 한다. 조건·기한부 승인제도는 재생의료제품의 특성을 반영한 일본 고유의 제도로, 대규모 장기 임상시험이 현실적으로 어려운 희귀질환 또는 생명을 위협하는 질환을 대상으로 한다.
해당 제도에서는 제한적 임상자료를 근거로 유효성이 예측되고 안전성이 확인된 경우 조건부 허가가 가능하다. 다만, 허가 시점에 사후 임상계획 제출이 요구되며, 정해진 기간 내에 유효성을 입증하지 못할 경우 허가는 효력을 상실한다.
최근에는 조건·기한부 승인제도의 예측 가능성을 높이기 위해 가이드라인이 개정·발표됐다. 해당 지침은 상담 및 심사 경험을 축적한 내용을 반영해 절차와 요구자료를 구체화하고 있다.
특히 약물 접근 지연(Drug lag, Drug loss) 문제를 최소화하기 위해 희귀질환 재생의료제품의 경우 미국이나 유럽에서 승인된 제품에 대해 일본인 임상자료를 사전 허가 요건으로 반드시 요구하지 않을 수 있다는 유연성도 언급됐다. 다만, 허가 후 일본 환자 대상 자료를 수집해 유효성과 안전성을 확인해야 한다는 조건이 따른다.
재생의료제품의 임상시험계획 신고 건수는 연간 약 30건 수준이며, 과학적 자문은 연간 80~100건 수준으로 보고됐다. 임상시험 유형별로는 세포치료제가 절반가량을 차지하고, in vivo 및 ex vivo 유전자치료제가 각각 약 4분의 1 수준으로 나타났다.
국제조화 활동과 관련해서는 ICH, IPRP 등 다자간 협력체를 통한 재생의료제품 규제 프레임워크 논의가 진행 중이라고 설명했다.
특히 세포·유전자치료제의 비교성(comparability) 이슈와 미래 조화 전략 수립을 위한 논의가 이루어지고 있다. 또한 PMDA는 2016년 설립된 PMDA Asia Training Center(PMDA-ATC)를 통해 아시아 지역 규제조화를 지원하고 있으며, 방콕과 워싱턴 D.C.에 국제사무소를 개소해 정보 교류와 제품 개발 유치를 강화하고 있다.
PMDA는 승인된 모든 재생의료제품에 대해 영어 평가보고서를 공개하고 있으며, 일본 내 개발을 희망하는 기업을 위한 전용 정보 페이지도 운영 중이라고 밝혔다.
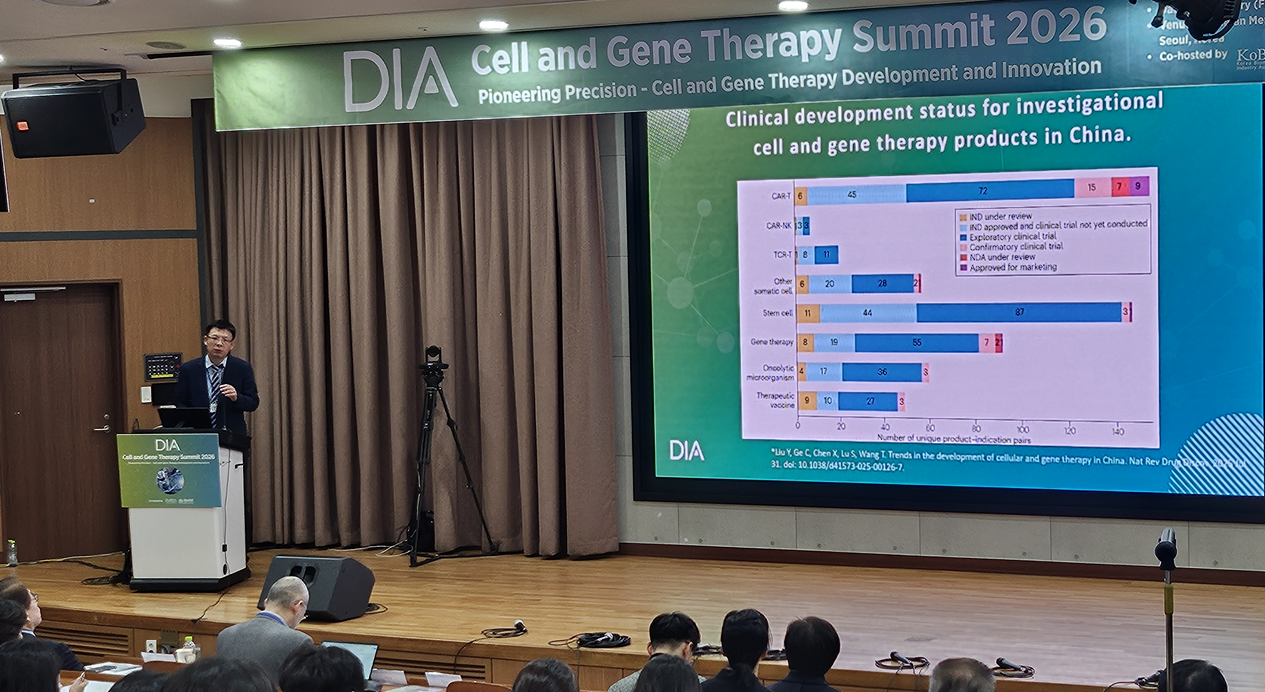
NMPA
Jinanchao Gao(Associate Researcher, Changping Laboratory)는 중국의 세포·유전자치료제 개발 현황과 국가약품감독관리국(NMPA)의 가속 심사체계, 조건부 승인 제도, 기술지침 운영 현황을 중심으로 발표했다.
발표에 따르면, 현재 중국에서는 다수의 세포치료제와 줄기세포 기반 제품, 유전자치료제가 승인됐으며, 그 외 종양용해바이러스(oncolytic virus), 치료용 백신 등 다양한 첨단치료제가 임상 개발 단계에 있다.
세포치료제의 적응증은 종양 분야 비중이 높고, 특히 혈액암과 고형암이 상당 부분을 차지하고 있다. 줄기세포 치료제는 류마티스질환, 신경계질환, 위장관질환, 호흡기질환 등 비암성 질환에서도 개발이 이뤄지고 있다. 유전자치료제는 주로 유전질환을 대상으로 개발되고 있다.
중국 내 임상연구 비중도 증가 추세에 있다. 최근 10년간 주요 의학저널에 게재된 중재적 임상시험 약 6만 건 중 중국이 차지하는 비중은 약 7% 수준이며, 세포·유전자치료제 분야에서는 약 10% 수준으로 더 높게 나타났다. 특히 암 적응증 비율은 글로벌 평균 대비 중국이 더 높은 것으로 보고됐다.
NMPA는 혁신의약품 개발을 촉진하기 위해 다양한 가속 프로그램을 운영하고 있다. 기업은 개발 초기 단계에서 기술적 쟁점에 대해 당국과 사전 소통을 진행할 수 있으며, 임상시험계획 신청은 원칙적으로 60일 내 묵시적 승인(implied license) 방식으로 처리된다. 또한 개발 과정 전반에서 보완자료 제출과 추가 소통이 가능하다.
가속 제도로는 혁신치료제 지정(Breakthrough Therapy Designation), 우선심사(Priority Review and Approval), 조건부 승인(Conditional Approval) 등이 운영된다. 혁신치료제 지정은 생명을 위협하는 질환 또는 중대한 삶의 질 저하를 초래하는 질환을 대상으로 하며, 기존 치료법이 없거나 현저한 임상적 이점을 입증한 경우 신청 가능하다. 지정 시 자원 우선 배정, 강화된 소통 및 개발 촉진 지원이 제공된다.
우선심사 대상에는 임상적으로 긴급한 의약품, 주요 감염병 및 희귀질환 치료를 위한 혁신의약품, 공중보건상 긴급성이 있는 백신 등이 포함된다. 일반 심사기간이 약 200일인 반면, 우선심사 지정 시 130일로 단축되며, 해외에서 이미 허가됐으나 중국에서 아직 시판되지 않은 긴급 의약품의 경우 70일까지 단축될 수 있다. 또한 기술검증, 시험·검사, 현장실사 등이 병행 처리된다.
조건부 승인 제도는 생명을 위협하는 질환이나 중대한 공중보건 수요가 있는 경우 적용된다. 임상시험에서 임상적 유익을 예측할 수 있는 대리지표(surrogate endpoint)나 조기 평가 가능한 중간지표(interim endpoint)를 근거로 허가가 가능하다. 다만 허가 신청 시 사후 임상계획을 함께 제출해야 하며, 정해진 기간 내 임상시험을 완료하고 최종보고서를 제출해야 한다. 사후 연구에서 유익성이 위험을 상회함이 입증되면 정식 승인으로 전환되지만, 이를 입증하지 못하거나 기한을 준수하지 못할 경우 허가 취소 등 행정조치가 가능하다.
중국은 해외 임상자료 활용에 대한 지침도 마련했다. 해외 임상시험 자료를 근거로 허가 신청이 가능하며, 필요 시 소규모 중국인 브리징 임상을 수행하는 방식이 활용된다.
또한 실제임상자료(real-world data)에 기반한 허가 신청을 지원하기 위한 지침도 발표됐다.규제과학 측면에서는 ‘중국 규제과학 행동계획(China Regulatory Science Action Plan)’을 통해 세포·유전자치료제 평가 방법과 기술체계를 정립하고 있다.
이와 함께 세포·유전자치료제의 임상시험 설계, 면역세포치료제, 종양용해바이러스, 유전자치료제, 줄기세포 제품, 임상위험관리계획 등과 관련한 다수의 기술지침이 발표됐다. 반복복제 바이러스 검출, 유전자변형 세포치료제의 비임상 평가, AAV 기반 유전자치료제 평가 등 구체적 요구사항도 정리돼 있다.
Jinanchao Gao는 중국이 환자 임상적 필요를 우선순위에 두고 세포·유전자치료제 개발을 가속화하기 위한 규제 프레임워크를 구축하고 있으며, 명확한 임상적 유익과 위험 자료를 기반으로 허가를 운영하고 있다고 설명하며 발표를 마무리했다.

식품의약품안전처
왕소영 식품의약품안전처 식품의약품안전평가원 세포유전자치료제과장은 국내 세포·유전자치료제(CGT) 규제체계, 허가 및 개발 현황, 가이드라인 운영 현황과 함께 최근 마련된 비교동등성 평가 가이드라인의 주요 내용을 발표했다.
발표에 따르면, 국내 첨단바이오의약품은 약사법과 첨단재생의료 및 첨단바이오의약품 안전 및 지원에 관한 법률에 따라 관리되고 있다. 기존 약사법이 화학의약품 중심으로 설계된 한계를 보완하기 위해 2019년 제정되어 2020년 시행된 첨단재생바이오법을 통해 세포·유전자치료제의 특성을 반영한 별도 관리체계를 구축했다. 해당 법 체계는 임상개발 단계부터 품목허가, 시판 후 안전관리까지 전주기 관리체계를 포함하고 있다.
국내에서는 첨단재생의료와 첨단바이오의약품을 구분해 운영하고 있다. 첨단재생의료는 보건복지부 주관 하에 연구 수행의 적절성을 중심으로 관리되며, 첨단바이오의약품은 전통적인 의약품 허가체계에 따라 품질·안전성·유효성 심사를 거쳐 허가된다. 세포·유전자치료제의 특성을 고려해 세포처리시설 관리, 인체세포등 관리업, 장기추적조사 등 별도 안전관리 체계도 운영 중이다.
2025년 기준 국내 허가된 세포치료제는 총 12개 품목으로, 줄기세포 치료제 4품목, 면역세포 치료제 1품목, 체세포 치료제 7품목이 포함된다. 다만 2019년 이후 신규 세포치료제 허가는 이루어지지 않은 상황이다. 유전자치료제는 총 6품목이 허가됐으며, CAR-T 치료제 3품목과 AAV 벡터 기반 치료제 3품목으로 구성돼 있다.
왕 과장은 주로 혈액암 등 항암제와 희귀질환 치료제가 중심을 이루고 있으며, 국내 개발 CAR-T 치료제에 대한 심사도 진행 중이라고 설명했다.
임상시험계획(IND) 승인 건수는 품목허가 대비 훨씬 많은 상황이다. 세포치료제 IND는 코로나19 이후 다소 감소 추세이나, 기존 MSC 중심에서 오가노이드, iPSC 등 새로운 유형의 세포치료제로 개발 방향이 다변화되고 있다. 유전자치료제는 표적화 기술 발전에 따라 IND 신청이 지속적으로 증가하고 있으며, CAR-T 외에도 다양한 벡터 및 유전자 전달기술을 활용한 품목이 확대되고 있다.
식약처는 제품화 지원을 위해 상담과 함께 가이드라인을 지속적으로 발간하고 있으며, 2025년 기준 운영 중인 가이드라인은 총 336개에 달한다. 기술 발전 속도를 반영해 연간 10건 이상 개정이 이뤄지고 있다.
이번 발표의 핵심은 2024년 마련된 세포·유전자치료제 비교동등성 평가 가이드라인이었다. 세포·유전자치료제는 개발 초기 소규모 제조에서 시작해 개발 단계가 진행될수록 제조공정 변경, 제조소 이전, 공정 조건 변경 등이 빈번하게 발생한다. 이에 따라 변경 전후 제품의 비교동등성 평가에 대한 질의와 보완 요구가 증가해 가이드라인을 마련했다는 설명이다.
왕 과장은 비교동등성의 개념이 ‘동일성’ 입증이 아니라, 변경으로 인해 안전성·유효성에 임상적으로 의미 있는 차이가 없는지를 과학적으로 입증하는 과정이라고 강조했다. 단순히 변경 전후 제품이 각각 품질기준에 적합하다는 사실만으로는 충분하지 않으며, 제품의 주요 품질특성(CQA)을 기반으로 위험도 평가를 수행해야 한다고 설명했다.
위해도 기반 접근을 통해 변경이 안전성·유효성에 미치는 영향 가능성을 분석하고, 영향이 클 것으로 예상되는 요소는 엄격한 통계적 분석과 충분한 배치 수 확보를 통해 평가해야 한다고도 밝혔다. 반면 영향이 상대적으로 적은 요소는 과학적 근거에 따라 유연한 범위 설정이 가능하다. 품질 데이터뿐 아니라 비임상 및 임상 데이터를 종합적으로 고려해 판단해야 하며, 가능하면 주요 제조 변경은 후기 임상 단계 이전에 완료하는 것이 권고된다고 덧붙였다.
분석 전략 측면에서는 단일 시험에 의존하기보다 상호보완적인 분석법을 활용하고, 가능한 한 민감한 시험법을 적용할 것을 권고했다. 연구 설계 단계에서는 물리·화학적 특성, 생물학적 특성, 공정 밸리데이션 자료 등을 종합적으로 검토해야 하며, 자가세포치료제의 경우 동일 공여자 검체를 분할해 비교하는 접근도 권고되고 있다.
제조공정 변경 이후에는 불순물, 공정유래 오염물질, 세포은행 변경 여부, 바이러스 안전성 등 추가 평가가 필요할 수 있으며, 품질 변화만으로 임상적 영향 예측이 어려운 경우에는 비임상 또는 임상시험이 추가로 요구될 가능성도 있다. 또한 세포 유형 변경, 벡터 세로타입 변경, 신규 유전자 도입 등은 경우에 따라 단순 변경이 아닌 새로운 품목으로 판단될 수 있어 개별 사안별 검토가 필요하다고 설명했다.
비교동등성 평가는 시험결과의 단순 나열이 아니라, 변경 전 임상시험 결과의 외삽 가능성과 기존 근거의 연속성을 유지할 수 있는지를 판단하는 과정이라는 점도 재차 강조됐다. 제조 변경 발생 시 CQA 영향 분석, 위험도 평가, 단계적 검증을 거쳐 임상적으로 의미 있는 차이가 없는지를 종합 판단하는 체계적 접근이 필요하다고 정리했다.
왕 과장은 세포·유전자치료제 개발 과정에서 불가피한 제조 변경을 과학적·위해도 기반으로 관리함으로써, 제품 특성을 유지하면서도 임상 근거의 연속성을 확보하는 것이 규제의 핵심이라고 설명하며 발표를 마무리했다.





|
|
서울시 서초구 서초대로 115 정다운빌딩 4층 TEL : 02-3270-0114 FAX : 02-3270-0189 E-mail : webmaster@yakup.com |
발행일 : 1954. 03. 29 등록일 : 2016. 05. 02 대표이사 : 함태원 발행 · 편집인 : 함태원 등록번호 : 서울,아04071 |
사업자등록번호 : 106-81-10940 통신사업자신고번호 : 제2022-서울서초-2586호 청소년보호책임자 : 이종운
Copyright © Yakup.com All rights reserved.
|

- 최윤수 기자 jjysc0229@yakup.com
- 입력 2026.03.03 06:00 수정 2026.03.03 06:01
세포·유전자치료제(Cell & Gene Therapy, CGT)가 글로벌 의약품 개발의 핵심 축으로 부상하면서, 각국 규제기관은 기존 의약품 규제 틀을 유지하면서도 첨단 치료제의 특성을 반영한 제도적 진화를 동시에 모색하고 있다. 생물학적 복잡성, 맞춤형 제조공정, 소규모·희귀질환 환자군 대상 임상 설계, 장기추적 안전성 관리 등 CGT 고유의 특성은 허가 심사, 사후 관리, 국제 조화 전략 전반에 걸쳐 새로운 기준 정립을 요구하고 있다. 이에 따라 주요 규제기관들은 심사체계 단순화, 가속 승인 프로그램 확대, 위험 기반 접근, 비교성·비교동등성 평가 고도화, 실제임상자료 활용 등 다양한 정책 수단을 통해 개발 촉진과 환자 보호 간 균형을 재정립하는 방향으로 제도를 발전시키고 있다.
이 같은 흐름을 한 자리에서 조망하는 자리가 마련됐다. Drug Information Association(DIA)는 지난 27일 서울 세브란스병원 유일한 기념홀에서 ‘Pioneering Precision: Cell and Gene Therapy Development and Innovation’을 주제로 ‘DIA Cell and Gene Therapy Summit’을 개최했다.
이날 세션1 ‘Regulatory Overview of Cell and Gene Therapy Field’에서는 ▲Caroline Pothet, Advanced Therapies and Haemato-Oncology Head(European Medicines Agency(EMA)), ▲Tamei Elliott, DIA US Director of Global Scientific Content(U.S. Food and Drug Administration(FDA)), ▲Yasuhiro Kishioka(Review Director, Office of Cellular and Tissue-based Products(Pharmaceuticals and Medical Devices Agency(PMDA)), ▲Jinanchao Gao, Associate Researcher Changping Laboratory(National Medical Products Administration(NMPA)), ▲왕소영 식품의약품안전처 식품의약품안전평가원 세포유전자치료제과장(식품의약품안전처) 등 주요 규제기관 관계자들이 참여해 자국의 CGT 규제체계 현황과 최근 제도 개편 동향을 발표했다.
발표는 유럽의 ATMP 법제 개편과 위원회 구조 단순화, 미국의 위험 기반·생애주기 중심 심사 접근, 일본의 조건·기한부 승인제도와 이원화된 법체계, 중국의 가속 승인 프로그램과 조건부 허가 운영, 한국의 첨단재생바이오법 기반 전주기 관리체계와 비교동등성 평가 가이드라인 등으로 이어졌다.
각 기관은 허가 심사 효율화와 조기 접근성 확보, 제조 변경에 따른 비교성 관리, 사후 임상근거 축적, 국제 조화 활동 등 공통 과제를 공유하면서도, 자국 제도에 기반한 차별화된 정책 도구를 제시했다. 이번 세션은 CGT 시대를 맞아 글로벌 규제 환경이 어떠한 방향으로 정비되고 있는지를 종합적으로 보여주는 자리로 마련됐다.
EMA
Caroline Pothet, 유럽의약품청(EMA) Advanced Therapies and Haemato-Oncology Head는 ‘Regulatory Update from the EMA’를 주제로 유럽 첨단치료제(ATMP) 규제 체계의 현황과 향후 개편 방향을 설명했다.
발표에 따르면, 유럽 ATMP 규제의 근간은 2007년 제정돼 2009년 발효된 ATMP Regulation이다. 이 법은 특정 제품군에 적용되는 특별법(Lex specialis)으로, ATMP의 정의와 범위를 명확히 하고 개발사가 자사 제품의 ATMP 해당 여부에 대해 사전 분류(classification)를 요청할 수 있는 제도를 도입했다.
또한 첨단치료제위원회(CAT)를 설치하고 ATMP에 대해 중앙집중 허가절차(centralised procedure)를 의무화했으며, 품질·유효성·안전성 및 긍정적 이익-위험 평가 입증을 허가 요건으로 규정했다. 허가 후 추가 유효성 및 안전성 자료 제출 요구 근거도 마련했으며, 중소기업을 위한 ATMP 인증(certification) 제도도 포함했다.
ATMP는 크게 ▲유전자치료제(Gene Therapy Medicinal Products) ▲체세포치료제(Somatic Cell Therapy Medicinal Products) ▲조직공학제제(Tissue Engineered Products) ▲의료기기와 결합된 복합 ATMP로 구분된다.
유전자치료제는 재조합 핵산을 포함해 유전적 변화를 유도하는 제품이며, 체세포치료제와 조직공학제제는 실질적 조작(substantial manipulation)을 거친 세포·조직을 활용해 질환 치료 또는 조직 재생·대체를 목표로 한다.
Caroline Pothet은 2023년 12월 유럽연합 집행위원회, 이사회, 유럽의회가 정치적 합의에 도달한 EU 신 의약품 법제(New Pharmaceutical Legislation, NPL)에 대해서도 설명했다. 해당 법제는 2028년부터 적용될 예정이며, 환자 접근성 강화, 경쟁력 및 혁신 지원, 미래 위기 대비, 의약품 공급 안정화 등 네 가지 축을 중심으로 구성된다.
ATMP와 관련해 가장 주목되는 변화는 유전자치료제 정의의 확대다. 기존 ‘recombinant nucleic acid’ 중심 정의에서 ‘synthetic nucleic acid’까지 범위를 넓히고, 유전자편집(gene editing) 기능을 명시적으로 포함할 예정이다.
허가 체계 측면에서는 위원회 구조가 단순화된다. 현재 CAT, CHMP, PRAC, PDCO, COMP 등 복수 위원회가 관여하는 구조에서 향후 CHMP와 PRAC 중심의 체계로 재편된다. 이에 따라 ATMP도 CHMP가 직접 평가를 담당하며, PRAC은 안전성 및 약물감시를 지속적으로 담당한다. 소아개발계획(PIP) 및 희귀의약품 관련 기능은 EMA와 CHMP가 분담하는 구조로 조정된다.
평가기간도 기존 210일에서 180일로 단축되며, 허가 갱신 절차 없이 무기한 허가가 부여된다. 또한 전자 제출 의무화, 임상시험 데이터의 전자적 제출, 전자 제품정보(electronic product information) 도입 등이 포함된다.
임상적 가치 평가와 관련해서는 2025년 1월부터 ATMP 및 항암제를 대상으로 EU 차원의 공동 임상평가(Joint Clinical Assessment, JCA)가 의무화됐다. 이는 허가 심사와 병행해 HTA 기관과의 조율을 진행하는 구조로, 이후 적용 범위가 점진적으로 확대될 예정이다.
ATMP Regulation의 특징 중 하나인 병원면제(Hospital Exemption) 제도에 대해서도 개편이 예고됐다. 국가별 관리 편차를 줄이기 위해 병원면제 제품의 공공 등록 의무화, 사용 의사의 유효성·안전성 데이터 수집 의무, 회원국 감독 강화 등이 도입될 예정이다.
혁신 지원 측면에서는 과학자문 체계 확대, HTA 기관과의 공동 절차 도입, PRIME 제도의 법제화 및 조기 지원 강화, 규제 샌드박스(Regulatory Sandbox) 도입, 소아개발계획의 연속적 업데이트 허용 등이 포함된다.
또한 ATMP 분류 제도는 유지되며, 향후에는 해당 제품이 의약품에 해당하는지 여부에 대해서도 EMA가 과학적 의견을 제시할 수 있게 된다. 의료기기·식품 등 타 규제 체계와의 협의도 가능하도록 구조가 조정된다.
Caroline Pothet은 이번 개편이 규제 간소화와 절차 효율화, 유전자치료 정의 확대, 병원면제 감독 강화, 디지털 전환 가속화 등을 포함하는 전면적 구조 개편이라고 설명하며 발표를 마무리했다.
FDA
Tamei Elliott(DIA US Director of Global Scientific Content)는 ‘FDA Update’를 주제로 미국 식품의약국(FDA)의 세포·유전자치료제 규제 체계와 최근 정책 방향을 설명했다.
발표에 따르면, 세포·유전자치료제의 기술적 특성과 개발 환경 변화는 규제 과학의 진화를 촉진하고 있다. 플랫폼 기술의 빠른 발전, 소규모·희귀질환 환자군을 대상으로 한 치료제 개발 확대, 복잡한 제조공정과 빈번한 생애주기 변경, 장기 안전성 모니터링 의무 등은 기존 규제 접근 방식에 보완을 요구하고 있다는 설명이다. 이에 따라 FDA는 혁신 촉진과 환자 보호 사이의 균형을 유지하는 방향으로 규제 적용 방식을 조정하고 있다고 밝혔다.
미국에서 대부분의 세포·유전자치료제는 공중보건서비스법(Public Health Service Act) 제351조에 따른 생물학적 제제로 규제된다. 이들 제품은 안전성, 순도, 효력에 대한 전면적 사전 심사를 거쳐야 하며, 개발은 임상시험계획(IND) 제출로 시작해 임상시험을 진행한 후 생물학적제제허가신청(BLA)으로 이어진다.
Tamei Elliott은 법적 기반과 핵심 요건 자체는 유지되고 있으며, 세포·유전자치료제라고 해서 품질·안전성·유효성에 대한 기본 의무가 완화되는 것은 아니라고 설명했다.
다만 FDA는 이러한 기존 규정을 세포·유전자치료제의 특성에 맞게 적용하는 방식에서 진화를 보이고 있다. 근거 창출 전략, 제조 및 품질관리(CMC), 비교성 관리, 효력 시험 전략 등에서 기대 수준이 구체화되고 있으며, 특히 생애주기 관점에서의 사전 계획을 강조하고 있다. 제조 변경이나 공정 개선을 개발 후반에 대응적으로 처리하기보다, 초기 단계부터 비교성 전략을 설계할 것을 요구하고 있다는 설명이다.
FDA는 세포·유전자치료제와 관련한 다양한 가이드라인을 제시하고 있으며, 이는 임상개발, 첨단 치료기술, CMC 및 품질, 프로그램 정책 등을 포괄한다. 가이드라인은 법적 구속력을 갖는 규정은 아니지만, 해당 분야에 대한 FDA의 현재 사고방식을 반영하는 문서로 활용된다. 소규모 환자군을 대상으로 한 임상시험 설계와 관련해서는 단일군 시험에서 피험자를 자기 대조군으로 활용하는 방식, 질병 진행 모델링, 외부 대조군 연구, 적응적 설계, 베이지안 접근, 마스터 프로토콜 등 다양한 혁신적 설계 방안이 논의되고 있다.
허가 후 안전성 및 유효성 자료 수집과 관련해서는 실제임상자료 및 실제임상근거 활용, 환자등록시스템 구축, 전자의무기록, 의료 청구자료, 생명통계자료 등 다양한 데이터원을 활용하는 방안이 제시됐다. 이는 장기 추적관찰이 필요한 세포·유전자치료제의 특성을 반영한 접근이다.
발표에서는 중증 질환을 대상으로 하는 치료제에 적용 가능한 신속 프로그램도 소개됐다. Fast Track, Breakthrough Therapy, Regenerative Medicine Advanced Therapy(RMAT), Priority Review, Accelerated Approval 등이 포함되며, 재생의학 치료는 세포치료제, 조직공학 제품, 인체세포·조직 제품, 관련 복합제품 등으로 정의된다.
최근 FDA는 세포·유전자치료제에 대해 위험 기반·과학 기반 유연성을 강조하고 있다. 다만 유연성은 규제 수준의 완화를 의미하지 않으며, 제품 위험도와 과학적 근거에 따라 개별적으로 적용된다. 예로는 임상 2·3상 이전 단계에서 상업용 완전 cGMP 요건을 일괄적으로 요구하지 않는 점, 공정 및 방법 검증에 대한 생애주기 접근, 공정적격성평가(PPQ) 배치 수의 유연한 적용, 소규모 환자군 대상 출하 기준의 탄력적 설정, 제조 경험에 따른 허가 후 기준 조정 등이 제시됐다.
FDA는 개발 초기 단계에서의 조기 소통을 지속적으로 권장하고 있으며, Pre-IND, End-of-Phase 2, Pre-Phase 3, BLA 단계 등에서 공식 회의(Type meeting)를 운영하고 있다. 서면 요청 시 60일 이내 회의를 개최해야 하는 절차가 마련돼 있다. 또한 라운드테이블과 워크숍을 통해 개발 과정의 도전과제를 파악하고 이를 향후 가이드라인과 심사 접근에 반영하고 있다고 설명했다.
최근 심사 환경과 관련해서는 문서화 수준 강화, 비교성 전략 및 평가변수의 타당성 확보, 비공식적 합의에 의존하지 않는 전략 수립의 필요성이 언급됐다.
Tamei Elliott은 세포·유전자치료제 개발 시 FDA의 생애주기 기대 수준을 반영해 초기 단계부터 규제 전략을 설계해야 하며, 가이드라인의 변화는 향후 심사 방향을 가늠할 수 있는 신호로 해석할 필요가 있다고 정리했다.
PMDA
Yasuhiro Kishioka(Review Director, Office of Cellular and Tissue-based Products, PMDA)는 일본의 재생의료제품 규제체계와 최근 승인 동향, 개발 촉진 제도 및 국제조화 활동을 중심으로 발표했다.
발표에 따르면, 일본은 2014년 재생의료에 대한 기대 증가를 배경으로 두 개의 법 체계를 정비했다. 하나는 ‘재생의료 등의 안전성 확보 등에 관한 법률(Safety Act)’이며, 다른 하나는 약사법을 개정한 ‘의약품의료기기등법(PMD Act)’이다.
Safety Act는 의료기관에서 시행되는 의료행위 및 비상업적 임상연구를 규율하며, 후생노동성이 관할한다. 반면 PMD Act는 재생의료제품의 임상시험계획 신고, 허가 심사 및 과학적 자문을 포함한 규제 절차를 규율하며, PMDA가 심사를 담당한다.
PMD Act상 재생의료제품은 세포기반 제품, 조직기반 제품, 유전자치료제를 포함하는 개념으로 정의된다.
Yasuhiro Kishioka는 ATMP와 유사한 범주로 이해할 수 있으나, 일본 법체계에 근거한 독자적 정의가 적용된다고 설명했다.
현재 일본에서 PMD Act에 따라 승인된 재생의료제품은 다수에 이르며, 적응증별로는 종양 분야 제품 비중이 높은 것으로 나타났다. 체세포치료제와 줄기세포 기반 제품도 다양한 적응증에서 허가를 받았다. 최근에는 iPS 세포 유래 제품 2건이 후생노동성 자문기구에서 조건부 승인을 권고받았다고 언급됐다.
일본은 재생의료제품 개발 촉진을 위해 오픈지정제도(Orphan designation), 사키가케(Sakigake) 지정, 조건·기한부 승인제도(Conditional and Time-limited Approval) 등 세 가지 제도를 운영하고 있다. 전체 승인 제품 중 80% 이상이 희귀의약품으로 지정됐으며, 지정 시 우선 과학적 자문, 우선 심사 등의 인센티브가 제공된다. 일반적인 허가 심사기간은 12개월이지만, 희귀 지정 제품의 경우 9개월로 단축된다.
사키가케 지정은 혁신 제품의 조기 도입을 목적으로 하며, 일본에서 최초 또는 글로벌과 동시 개발을 추진하는 경우를 요건으로 한다. 조건·기한부 승인제도는 재생의료제품의 특성을 반영한 일본 고유의 제도로, 대규모 장기 임상시험이 현실적으로 어려운 희귀질환 또는 생명을 위협하는 질환을 대상으로 한다.
해당 제도에서는 제한적 임상자료를 근거로 유효성이 예측되고 안전성이 확인된 경우 조건부 허가가 가능하다. 다만, 허가 시점에 사후 임상계획 제출이 요구되며, 정해진 기간 내에 유효성을 입증하지 못할 경우 허가는 효력을 상실한다.
최근에는 조건·기한부 승인제도의 예측 가능성을 높이기 위해 가이드라인이 개정·발표됐다. 해당 지침은 상담 및 심사 경험을 축적한 내용을 반영해 절차와 요구자료를 구체화하고 있다.
특히 약물 접근 지연(Drug lag, Drug loss) 문제를 최소화하기 위해 희귀질환 재생의료제품의 경우 미국이나 유럽에서 승인된 제품에 대해 일본인 임상자료를 사전 허가 요건으로 반드시 요구하지 않을 수 있다는 유연성도 언급됐다. 다만, 허가 후 일본 환자 대상 자료를 수집해 유효성과 안전성을 확인해야 한다는 조건이 따른다.
재생의료제품의 임상시험계획 신고 건수는 연간 약 30건 수준이며, 과학적 자문은 연간 80~100건 수준으로 보고됐다. 임상시험 유형별로는 세포치료제가 절반가량을 차지하고, in vivo 및 ex vivo 유전자치료제가 각각 약 4분의 1 수준으로 나타났다.
국제조화 활동과 관련해서는 ICH, IPRP 등 다자간 협력체를 통한 재생의료제품 규제 프레임워크 논의가 진행 중이라고 설명했다.
특히 세포·유전자치료제의 비교성(comparability) 이슈와 미래 조화 전략 수립을 위한 논의가 이루어지고 있다. 또한 PMDA는 2016년 설립된 PMDA Asia Training Center(PMDA-ATC)를 통해 아시아 지역 규제조화를 지원하고 있으며, 방콕과 워싱턴 D.C.에 국제사무소를 개소해 정보 교류와 제품 개발 유치를 강화하고 있다.
PMDA는 승인된 모든 재생의료제품에 대해 영어 평가보고서를 공개하고 있으며, 일본 내 개발을 희망하는 기업을 위한 전용 정보 페이지도 운영 중이라고 밝혔다.
NMPA
Jinanchao Gao(Associate Researcher, Changping Laboratory)는 중국의 세포·유전자치료제 개발 현황과 국가약품감독관리국(NMPA)의 가속 심사체계, 조건부 승인 제도, 기술지침 운영 현황을 중심으로 발표했다.
발표에 따르면, 현재 중국에서는 다수의 세포치료제와 줄기세포 기반 제품, 유전자치료제가 승인됐으며, 그 외 종양용해바이러스(oncolytic virus), 치료용 백신 등 다양한 첨단치료제가 임상 개발 단계에 있다.
세포치료제의 적응증은 종양 분야 비중이 높고, 특히 혈액암과 고형암이 상당 부분을 차지하고 있다. 줄기세포 치료제는 류마티스질환, 신경계질환, 위장관질환, 호흡기질환 등 비암성 질환에서도 개발이 이뤄지고 있다. 유전자치료제는 주로 유전질환을 대상으로 개발되고 있다.
중국 내 임상연구 비중도 증가 추세에 있다. 최근 10년간 주요 의학저널에 게재된 중재적 임상시험 약 6만 건 중 중국이 차지하는 비중은 약 7% 수준이며, 세포·유전자치료제 분야에서는 약 10% 수준으로 더 높게 나타났다. 특히 암 적응증 비율은 글로벌 평균 대비 중국이 더 높은 것으로 보고됐다.
NMPA는 혁신의약품 개발을 촉진하기 위해 다양한 가속 프로그램을 운영하고 있다. 기업은 개발 초기 단계에서 기술적 쟁점에 대해 당국과 사전 소통을 진행할 수 있으며, 임상시험계획 신청은 원칙적으로 60일 내 묵시적 승인(implied license) 방식으로 처리된다. 또한 개발 과정 전반에서 보완자료 제출과 추가 소통이 가능하다.
가속 제도로는 혁신치료제 지정(Breakthrough Therapy Designation), 우선심사(Priority Review and Approval), 조건부 승인(Conditional Approval) 등이 운영된다. 혁신치료제 지정은 생명을 위협하는 질환 또는 중대한 삶의 질 저하를 초래하는 질환을 대상으로 하며, 기존 치료법이 없거나 현저한 임상적 이점을 입증한 경우 신청 가능하다. 지정 시 자원 우선 배정, 강화된 소통 및 개발 촉진 지원이 제공된다.
우선심사 대상에는 임상적으로 긴급한 의약품, 주요 감염병 및 희귀질환 치료를 위한 혁신의약품, 공중보건상 긴급성이 있는 백신 등이 포함된다. 일반 심사기간이 약 200일인 반면, 우선심사 지정 시 130일로 단축되며, 해외에서 이미 허가됐으나 중국에서 아직 시판되지 않은 긴급 의약품의 경우 70일까지 단축될 수 있다. 또한 기술검증, 시험·검사, 현장실사 등이 병행 처리된다.
조건부 승인 제도는 생명을 위협하는 질환이나 중대한 공중보건 수요가 있는 경우 적용된다. 임상시험에서 임상적 유익을 예측할 수 있는 대리지표(surrogate endpoint)나 조기 평가 가능한 중간지표(interim endpoint)를 근거로 허가가 가능하다. 다만 허가 신청 시 사후 임상계획을 함께 제출해야 하며, 정해진 기간 내 임상시험을 완료하고 최종보고서를 제출해야 한다. 사후 연구에서 유익성이 위험을 상회함이 입증되면 정식 승인으로 전환되지만, 이를 입증하지 못하거나 기한을 준수하지 못할 경우 허가 취소 등 행정조치가 가능하다.
중국은 해외 임상자료 활용에 대한 지침도 마련했다. 해외 임상시험 자료를 근거로 허가 신청이 가능하며, 필요 시 소규모 중국인 브리징 임상을 수행하는 방식이 활용된다.
또한 실제임상자료(real-world data)에 기반한 허가 신청을 지원하기 위한 지침도 발표됐다.규제과학 측면에서는 ‘중국 규제과학 행동계획(China Regulatory Science Action Plan)’을 통해 세포·유전자치료제 평가 방법과 기술체계를 정립하고 있다.
이와 함께 세포·유전자치료제의 임상시험 설계, 면역세포치료제, 종양용해바이러스, 유전자치료제, 줄기세포 제품, 임상위험관리계획 등과 관련한 다수의 기술지침이 발표됐다. 반복복제 바이러스 검출, 유전자변형 세포치료제의 비임상 평가, AAV 기반 유전자치료제 평가 등 구체적 요구사항도 정리돼 있다.
Jinanchao Gao는 중국이 환자 임상적 필요를 우선순위에 두고 세포·유전자치료제 개발을 가속화하기 위한 규제 프레임워크를 구축하고 있으며, 명확한 임상적 유익과 위험 자료를 기반으로 허가를 운영하고 있다고 설명하며 발표를 마무리했다.
식품의약품안전처
왕소영 식품의약품안전처 식품의약품안전평가원 세포유전자치료제과장은 국내 세포·유전자치료제(CGT) 규제체계, 허가 및 개발 현황, 가이드라인 운영 현황과 함께 최근 마련된 비교동등성 평가 가이드라인의 주요 내용을 발표했다.
발표에 따르면, 국내 첨단바이오의약품은 약사법과 첨단재생의료 및 첨단바이오의약품 안전 및 지원에 관한 법률에 따라 관리되고 있다. 기존 약사법이 화학의약품 중심으로 설계된 한계를 보완하기 위해 2019년 제정되어 2020년 시행된 첨단재생바이오법을 통해 세포·유전자치료제의 특성을 반영한 별도 관리체계를 구축했다. 해당 법 체계는 임상개발 단계부터 품목허가, 시판 후 안전관리까지 전주기 관리체계를 포함하고 있다.
국내에서는 첨단재생의료와 첨단바이오의약품을 구분해 운영하고 있다. 첨단재생의료는 보건복지부 주관 하에 연구 수행의 적절성을 중심으로 관리되며, 첨단바이오의약품은 전통적인 의약품 허가체계에 따라 품질·안전성·유효성 심사를 거쳐 허가된다. 세포·유전자치료제의 특성을 고려해 세포처리시설 관리, 인체세포등 관리업, 장기추적조사 등 별도 안전관리 체계도 운영 중이다.
2025년 기준 국내 허가된 세포치료제는 총 12개 품목으로, 줄기세포 치료제 4품목, 면역세포 치료제 1품목, 체세포 치료제 7품목이 포함된다. 다만 2019년 이후 신규 세포치료제 허가는 이루어지지 않은 상황이다. 유전자치료제는 총 6품목이 허가됐으며, CAR-T 치료제 3품목과 AAV 벡터 기반 치료제 3품목으로 구성돼 있다.
왕 과장은 주로 혈액암 등 항암제와 희귀질환 치료제가 중심을 이루고 있으며, 국내 개발 CAR-T 치료제에 대한 심사도 진행 중이라고 설명했다.
임상시험계획(IND) 승인 건수는 품목허가 대비 훨씬 많은 상황이다. 세포치료제 IND는 코로나19 이후 다소 감소 추세이나, 기존 MSC 중심에서 오가노이드, iPSC 등 새로운 유형의 세포치료제로 개발 방향이 다변화되고 있다. 유전자치료제는 표적화 기술 발전에 따라 IND 신청이 지속적으로 증가하고 있으며, CAR-T 외에도 다양한 벡터 및 유전자 전달기술을 활용한 품목이 확대되고 있다.
식약처는 제품화 지원을 위해 상담과 함께 가이드라인을 지속적으로 발간하고 있으며, 2025년 기준 운영 중인 가이드라인은 총 336개에 달한다. 기술 발전 속도를 반영해 연간 10건 이상 개정이 이뤄지고 있다.
이번 발표의 핵심은 2024년 마련된 세포·유전자치료제 비교동등성 평가 가이드라인이었다. 세포·유전자치료제는 개발 초기 소규모 제조에서 시작해 개발 단계가 진행될수록 제조공정 변경, 제조소 이전, 공정 조건 변경 등이 빈번하게 발생한다. 이에 따라 변경 전후 제품의 비교동등성 평가에 대한 질의와 보완 요구가 증가해 가이드라인을 마련했다는 설명이다.
왕 과장은 비교동등성의 개념이 ‘동일성’ 입증이 아니라, 변경으로 인해 안전성·유효성에 임상적으로 의미 있는 차이가 없는지를 과학적으로 입증하는 과정이라고 강조했다. 단순히 변경 전후 제품이 각각 품질기준에 적합하다는 사실만으로는 충분하지 않으며, 제품의 주요 품질특성(CQA)을 기반으로 위험도 평가를 수행해야 한다고 설명했다.
위해도 기반 접근을 통해 변경이 안전성·유효성에 미치는 영향 가능성을 분석하고, 영향이 클 것으로 예상되는 요소는 엄격한 통계적 분석과 충분한 배치 수 확보를 통해 평가해야 한다고도 밝혔다. 반면 영향이 상대적으로 적은 요소는 과학적 근거에 따라 유연한 범위 설정이 가능하다. 품질 데이터뿐 아니라 비임상 및 임상 데이터를 종합적으로 고려해 판단해야 하며, 가능하면 주요 제조 변경은 후기 임상 단계 이전에 완료하는 것이 권고된다고 덧붙였다.
분석 전략 측면에서는 단일 시험에 의존하기보다 상호보완적인 분석법을 활용하고, 가능한 한 민감한 시험법을 적용할 것을 권고했다. 연구 설계 단계에서는 물리·화학적 특성, 생물학적 특성, 공정 밸리데이션 자료 등을 종합적으로 검토해야 하며, 자가세포치료제의 경우 동일 공여자 검체를 분할해 비교하는 접근도 권고되고 있다.
제조공정 변경 이후에는 불순물, 공정유래 오염물질, 세포은행 변경 여부, 바이러스 안전성 등 추가 평가가 필요할 수 있으며, 품질 변화만으로 임상적 영향 예측이 어려운 경우에는 비임상 또는 임상시험이 추가로 요구될 가능성도 있다. 또한 세포 유형 변경, 벡터 세로타입 변경, 신규 유전자 도입 등은 경우에 따라 단순 변경이 아닌 새로운 품목으로 판단될 수 있어 개별 사안별 검토가 필요하다고 설명했다.
비교동등성 평가는 시험결과의 단순 나열이 아니라, 변경 전 임상시험 결과의 외삽 가능성과 기존 근거의 연속성을 유지할 수 있는지를 판단하는 과정이라는 점도 재차 강조됐다. 제조 변경 발생 시 CQA 영향 분석, 위험도 평가, 단계적 검증을 거쳐 임상적으로 의미 있는 차이가 없는지를 종합 판단하는 체계적 접근이 필요하다고 정리했다.
왕 과장은 세포·유전자치료제 개발 과정에서 불가피한 제조 변경을 과학적·위해도 기반으로 관리함으로써, 제품 특성을 유지하면서도 임상 근거의 연속성을 확보하는 것이 규제의 핵심이라고 설명하며 발표를 마무리했다.
무단 전재·복사·배포 등을 금지합니다.