

“제약사의 미국 진출, FDA 자원 활용이 큰 도움”
‘TPP 통한 목표 수립’ 및 ‘성공적인 Pre-IND 미팅’ 강조
 기자가 쓴 기사 더보기
기자가 쓴 기사 더보기
입력 2018.04.06 06:31
수정 2018.04.06 06:51




 많은 국내 제약사들이 미국 허가 관문을 뚫기 위해서는 TPP를 통해 임상 실패율을 줄이고 Pre-IND 미팅을 성공적으로 마칠 수 있도록 준비해야 할 것으로 전망됐다.
많은 국내 제약사들이 미국 허가 관문을 뚫기 위해서는 TPP를 통해 임상 실패율을 줄이고 Pre-IND 미팅을 성공적으로 마칠 수 있도록 준비해야 할 것으로 전망됐다.지난 5일 열린 서울대학교병원 임상시험센터 심포지엄에서는 한국인 최초로 FDA 부국장을 역임한 안해영 박사(현 AhnBio Consulting 대표)가 ‘Are you ready for clinical trials in the US?’를 주제로 강의를 펼쳤다.
안 박사는 “FDA에 신약 승인 신청(NDA)을 할 때는 △의약품이 규정된 용도 안에서 안전하고 효과적인지 △질(quality)적인 면에서 승인받을 만한 품질인지에 대해 증명해야 하고 △약의 라벨링(labeling)이 적절한 지에 대해 고려해야 한다”고 말했다.
이어 “제약사에서 임상 3상을 끝낸 후 FDA에 허가 신청서를 제출하면 FDA에서는 이를 10~12달 리뷰하는 작업을 거친다. 그러나 이 작업에서 끝날 수도 있지만 승인 이후 추가적인 실험을 요구하는 경우도 있다”고 설명했다.
예를 들어 first-in-class를 개발할 경우라면 위약을 대조군으로 두어야 한다. 그러나 fisrt-in-class가 아닌 두 번째로 약을 개발한 회사의 경우 위약 대조군보다는 능동적인 대조군을 두어 실험한 자료를 요구할 수 있다. 이처럼 상황에 따라 FDA가 요구하는 방향은 달라질 수 있다는 것.
그는 성공적인 신약 개발을 위해서는 ‘Target Product Profile(TPP)’가 중요하다고 강조했다.
TPP란 임상시험 전단계(Pre-IND)부터 임상시험 전반에 걸쳐 약물의 라벨링을 어떻게 작성할 것인가를 목표로 하고, 그 목표에 따라 신약 개발을 계획하고 임상시험들을 수행하는 것이다.
안 박사는 “임상 3상 진입 전 한국 개발자들이 사용하는 TPP와, FDA가 생각하는 TPP는 근본적으로 다르다. 미국의 경우 신약을 개발한 후 마지막으로 라벨링을 어떻게 작성할 것인가가 가장 큰 목표다”고 전했다.
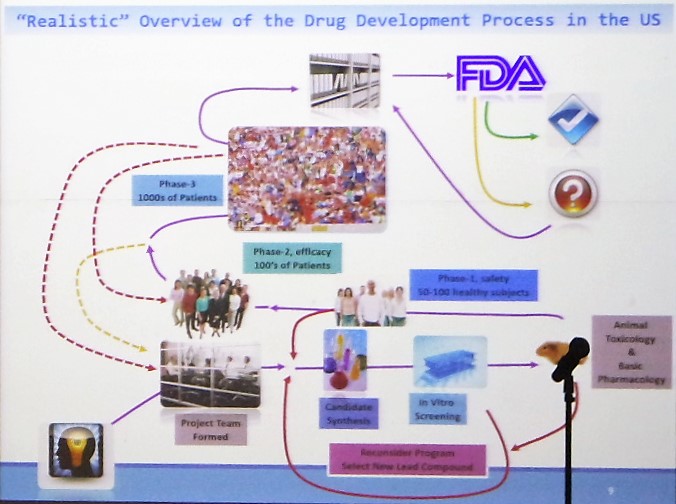
또 임상 2상과 3상에 들어가는 비용이 만만치 않은 만큼, 이 단계에서 실패를 줄이려면 처음부터 목표를 정확히 세우고 임상을 실시해야 한다고 강조했다.
안 박사는 “임상 진행 과정에서 CRO가 추가적인 실험을 요구할 경우 국내 제약사는 반박할 수 있는 여지가 많지 않다. 그러나 요구하는 모든 실험을 할 수도 없는 노릇이다. 따라서 정말 필요한 실험들만을 진행해서 승인 신청을 해야 한다. 이것의 바탕으로 TPP가 돼있어야만 가능하다”고 설명했다.
안 박사는 임상의 초기 단계부터 FDA와 상호작용할 것을 조언했다.
그는 “임상 3상이 끝나면 FDA에 신청서를 제출하는데, 이 때 FDA의 반응은 3가지다. △받아들일 수 있는(acceptable) 경우와 △승인 할 수 있는(approvable) 경우, △승인 할 수 없는(not approvable) 경우다”고 말했다.
이어 “임상이 어느 정도 진행되고, 비용은 비용대로 써버린 후에 미국 승인을 위해 FDA와 미팅을 가졌는데 알고 보니 FDA가 원하는 방향이 정반대라면 그것보다 낭패는 없다”며 “국내 제약사들은 임상 초기부터 FDA 스태프와 접촉해 미리 의견을 구해야 한다”고 말했다.
그는 “Pre-IND 미팅은 적어도 승인서 제출 예상 시점보다 최소 6개월 미리 신청해야 한다. Pre-IND 미팅은 1시간으로 시간이 제한돼 있고, 횟수 또한 1회만 가능한 규칙(Rule of Thumb)이 있다. 그러므로 이 한 번의 기회를 제대로 이용하려면 확실한 준비가 필요하다”고 강조했다.
이어 “미팅 과정에서 프레젠테이션을 발표할 수는 있지만, 그것 또한 1시간 안에서 이뤄져야 하기 때문에 프레젠테이션은 최대한 빨리 끝내고 토론하는 시간을 많이 가지는 것이 좋다. 또 급하게 추가된 신규 데이터는 주의 깊게 보지 않으므로 제출하지 말 것을 권한다”고 조언했다.
또 다가오는 5월 5일부터는 eCTD(전자공통기술문서) 양식을 이용해 FDA 내 인허가 기관인 CDER 및 CBER에 허가 신청서를 제출해야 한다고 덧붙였다.
안 박사는 이어 “Initial IND에서 △인체 피험자의 높은 부상 또는 질병 노출도 △임상 연구자의 자격 불충분 △IND 신청에 충분한 정보 불포함 △임상 관련 책자(investigator brochure)가 오해의 소지가 있거나 불완전한 경우가 있다면 FDA는 임상시험을 보류(clinical hold)하게 된다”며 해당 조건들을 면밀히 살필 것을 조언했다.
[관련기사]
안해영박사 “한국 제약·생명공학산업, 신약개발 변곡점”
2018-02-27 06:30
약업신문 공식 SNS 채널 구독






오늘의 헤드라인
전체댓글 0개
등록된 댓글이 없습니다.
| 인기기사 | 더보기 + |
-
1 상장 제약·바이오 1Q 평균 유보율 코스피 2324.16%·코스닥 2549.03% -
2 첫 국산 CAR-T 큐로셀의 '림카토', 남은 건 환자 접근성 -
3 “계약 종료도, 개발 중단도 아니다” 에이비엘 이상훈 대표, ABL301 우려 차단㊤ -
4 미국, 이번엔 빅파마 겨눴다…중국 임상·기술도입까지 국가안보 심사 확대 -
5 제약바이오 전시회 'CPHI/ Hi Korea', 역대 최대 규모 8월 25일 개막 -
6 이일형 변호사 “허가·특허·약가 얽힌 제약바이오, 법률 자문도 전략이 돼야” -
7 에이테크아이엔씨, 파마리서치서 ‘HA 필러 리쥬비엘’ 자동화 설비 수주 -
8 김혜진 KoNECT 신임 이사장 "AI·DCT 집중 지원…'세계 3대 임상 강국' 이끈다" -
9 에이비엘 이상훈 대표 “ABL001 허가 문턱, ABL111 3상 직행" 상업화 속도㊦ -
10 약사회 "동물병원 인체용 전문약 사용도 보고 의무화해야"
| 인터뷰 | 더보기 + |
| PEOPLE | 더보기 + |
| 컬쳐/클래시그널 | 더보기 + |
|
|
서울시 서초구 서초대로 115 정다운빌딩 4층 TEL : 02-3270-0114 FAX : 02-3270-0189 E-mail : webmaster@yakup.com |
발행일 : 1954. 03. 29 등록일 : 2016. 05. 02 대표이사 : 함태원 발행 · 편집인 : 함태원 등록번호 : 서울,아04071 |
사업자등록번호 : 106-81-10940 통신사업자신고번호 : 제2022-서울서초-2586호 청소년보호책임자 : 이종운
Copyright © Yakup.com All rights reserved.
|

“제약사의 미국 진출, FDA 자원 활용이 큰 도움”
‘TPP 통한 목표 수립’ 및 ‘성공적인 Pre-IND 미팅’ 강조
- 전세미 기자 jeonsm@yakup.com
- 입력 2018.04.06 06:31 수정 2018.04.06 06:51
 많은 국내 제약사들이 미국 허가 관문을 뚫기 위해서는 TPP를 통해 임상 실패율을 줄이고 Pre-IND 미팅을 성공적으로 마칠 수 있도록 준비해야 할 것으로 전망됐다.
많은 국내 제약사들이 미국 허가 관문을 뚫기 위해서는 TPP를 통해 임상 실패율을 줄이고 Pre-IND 미팅을 성공적으로 마칠 수 있도록 준비해야 할 것으로 전망됐다.지난 5일 열린 서울대학교병원 임상시험센터 심포지엄에서는 한국인 최초로 FDA 부국장을 역임한 안해영 박사(현 AhnBio Consulting 대표)가 ‘Are you ready for clinical trials in the US?’를 주제로 강의를 펼쳤다.
안 박사는 “FDA에 신약 승인 신청(NDA)을 할 때는 △의약품이 규정된 용도 안에서 안전하고 효과적인지 △질(quality)적인 면에서 승인받을 만한 품질인지에 대해 증명해야 하고 △약의 라벨링(labeling)이 적절한 지에 대해 고려해야 한다”고 말했다.
이어 “제약사에서 임상 3상을 끝낸 후 FDA에 허가 신청서를 제출하면 FDA에서는 이를 10~12달 리뷰하는 작업을 거친다. 그러나 이 작업에서 끝날 수도 있지만 승인 이후 추가적인 실험을 요구하는 경우도 있다”고 설명했다.
예를 들어 first-in-class를 개발할 경우라면 위약을 대조군으로 두어야 한다. 그러나 fisrt-in-class가 아닌 두 번째로 약을 개발한 회사의 경우 위약 대조군보다는 능동적인 대조군을 두어 실험한 자료를 요구할 수 있다. 이처럼 상황에 따라 FDA가 요구하는 방향은 달라질 수 있다는 것.
그는 성공적인 신약 개발을 위해서는 ‘Target Product Profile(TPP)’가 중요하다고 강조했다.
TPP란 임상시험 전단계(Pre-IND)부터 임상시험 전반에 걸쳐 약물의 라벨링을 어떻게 작성할 것인가를 목표로 하고, 그 목표에 따라 신약 개발을 계획하고 임상시험들을 수행하는 것이다.
안 박사는 “임상 3상 진입 전 한국 개발자들이 사용하는 TPP와, FDA가 생각하는 TPP는 근본적으로 다르다. 미국의 경우 신약을 개발한 후 마지막으로 라벨링을 어떻게 작성할 것인가가 가장 큰 목표다”고 전했다.

또 임상 2상과 3상에 들어가는 비용이 만만치 않은 만큼, 이 단계에서 실패를 줄이려면 처음부터 목표를 정확히 세우고 임상을 실시해야 한다고 강조했다.
안 박사는 “임상 진행 과정에서 CRO가 추가적인 실험을 요구할 경우 국내 제약사는 반박할 수 있는 여지가 많지 않다. 그러나 요구하는 모든 실험을 할 수도 없는 노릇이다. 따라서 정말 필요한 실험들만을 진행해서 승인 신청을 해야 한다. 이것의 바탕으로 TPP가 돼있어야만 가능하다”고 설명했다.
안 박사는 임상의 초기 단계부터 FDA와 상호작용할 것을 조언했다.
그는 “임상 3상이 끝나면 FDA에 신청서를 제출하는데, 이 때 FDA의 반응은 3가지다. △받아들일 수 있는(acceptable) 경우와 △승인 할 수 있는(approvable) 경우, △승인 할 수 없는(not approvable) 경우다”고 말했다.
이어 “임상이 어느 정도 진행되고, 비용은 비용대로 써버린 후에 미국 승인을 위해 FDA와 미팅을 가졌는데 알고 보니 FDA가 원하는 방향이 정반대라면 그것보다 낭패는 없다”며 “국내 제약사들은 임상 초기부터 FDA 스태프와 접촉해 미리 의견을 구해야 한다”고 말했다.
그는 “Pre-IND 미팅은 적어도 승인서 제출 예상 시점보다 최소 6개월 미리 신청해야 한다. Pre-IND 미팅은 1시간으로 시간이 제한돼 있고, 횟수 또한 1회만 가능한 규칙(Rule of Thumb)이 있다. 그러므로 이 한 번의 기회를 제대로 이용하려면 확실한 준비가 필요하다”고 강조했다.
이어 “미팅 과정에서 프레젠테이션을 발표할 수는 있지만, 그것 또한 1시간 안에서 이뤄져야 하기 때문에 프레젠테이션은 최대한 빨리 끝내고 토론하는 시간을 많이 가지는 것이 좋다. 또 급하게 추가된 신규 데이터는 주의 깊게 보지 않으므로 제출하지 말 것을 권한다”고 조언했다.
또 다가오는 5월 5일부터는 eCTD(전자공통기술문서) 양식을 이용해 FDA 내 인허가 기관인 CDER 및 CBER에 허가 신청서를 제출해야 한다고 덧붙였다.
안 박사는 이어 “Initial IND에서 △인체 피험자의 높은 부상 또는 질병 노출도 △임상 연구자의 자격 불충분 △IND 신청에 충분한 정보 불포함 △임상 관련 책자(investigator brochure)가 오해의 소지가 있거나 불완전한 경우가 있다면 FDA는 임상시험을 보류(clinical hold)하게 된다”며 해당 조건들을 면밀히 살필 것을 조언했다.
Copyright © Yakup.com All rights reserved.
약업신문 의 모든 컨텐츠(기사)는 저작권법의 보호를 받습니다.
무단 전재·복사·배포 등을 금지합니다.
무단 전재·복사·배포 등을 금지합니다.